A critical care doctor has said it’s a waste of taxpayers’ money for the U.S. government to purchase drug company Merck’s anti-viral treatment for COVID-19, if it’s approved by the Food and Drug Administration (FDA).
Dr. Pierre Kory, president and chief medical officer of the Front Line COVID-19 Critical Care Alliance (FLCCC), said there’s no reason for the Biden administration to “enrich Merck for a drug, which has already been the source of a whistleblower complaint, and which has already failed in hospitalized patients” when ivermectin, a “low cost, safe, widely available [drug that] has proven to work in many phases of the disease, not only as outpatients but inpatients” is ready to be distributed to Americans.

The drug’s potential to reduce the risk of death or hospitalization is being assessed in an ongoing phase three trial that plans to enroll 1,850 non-hospitalized patients with mild to moderate COVID-19. The final data is expected to be released as early as this fall.
However, a phase three trial to examine the drug in hospitalized patients was scrapped after findings from phase two showed molnupiravir was unlikely to demonstrate any clinical benefit.
Kory said Merck’s statement on ivermectin is untrue and hurts patients.
“I need to call attention to how disturbed I am at Merck,” Kory said, adding that the company “issued a statement that there was no evidence to support the efficacy of ivermectin.”
“They even called into question the safety of one of the safest drugs known in history. That statement was a lie. That was a lie, and it’s hurting patients and it’s caused an incalculable loss of life.”
Merck and the HHS did not respond to requests for comment.
Ivermectin is an FDA-approved anti-parasitic drug initially introduced for animals in 1981 before its widespread use in humans in 1987 to treat river blindness, lymphatic filariasis (a disease that causes severe swelling in the limbs), scabies, and head lice. It earned the title of wonder drug for its safety, efficacy, versatility, and global impact, with it being placed on the World Health Organization’s list of essential medicines.

The federal regulator didn’t reply to The Epoch Times’ inquiry about whether the EUA for all the COVID-19 vaccines would be terminated if molnupiravir was approved or issued emergency authorization.
Kory stated that areas that have adopted ivermectin into their COVID-19 treatment guidelines have seen a decrease in their COVID-19 case numbers and hospitalizations.
Mexico City implemented a home-care program to give out ivermectin kits to anyone who tested positive for COVID-19 and wanted to take the medication on Dec. 28, 2020, as the city dealt with rising cases and hospitalizations.
“Results showed up to 76 [percent] reduction in hospitalization in the group that was taking ivermectin,” according to the outlet.
If molnuvirapir gets approved by the FDA in the fall, the drug will be the second anti-viral treatment for COVID-19. Remdesivir was the first therapy approved in October 2020 for hospitalized patients.
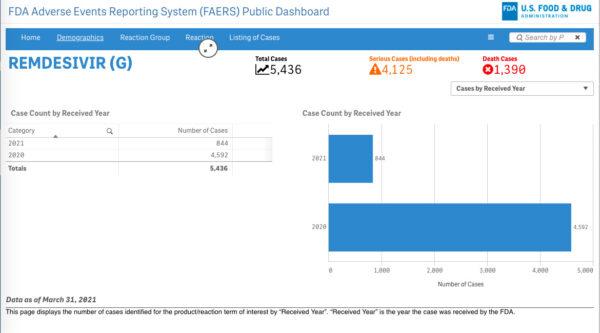
More than 4,000 reports of serious adverse events and 1,390 cases of deaths from remdesivir have been made on the FDA’s Adverse Events Reporting System (FAERS) as of March 31, 2021.
For ivermectin, about 2,311 serious adverse reactions and 373 cases of deaths were submitted to FAERS between 1996 to March 2021.
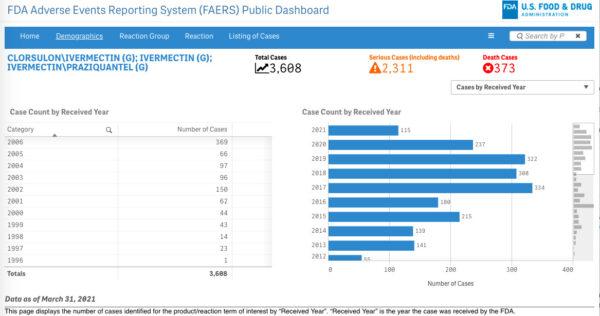
FAERS is a tool that allows users to search for “data on adverse events reported to the FDA for many drug and biologic products.”