SPECIAL COVERAGE
Read More




















Read More
Biden Signs $95 Billion Foreign Aid Bill Into Law
The package includes aid to Ukraine, Israel, and Taiwan, as well as a measure requiring TikTok’s Chinese parent company to divest the app.



When they thought all options were exhausted, precision medicine provides new hope with treatments as unique as their symptoms.
When they thought all options were exhausted, precision medicine provides new hope with treatments as unique as their symptoms.
Biden Signs $95 Billion Foreign Aid Bill Into Law
The package includes aid to Ukraine, Israel, and Taiwan, as well as a measure requiring TikTok’s Chinese parent company to divest the app.







Trending Videos

























Top Premium Reads










Top Stories









Most Read

Trump Named as Unindicted Co-Conspirator in Michigan ‘Fake Electors’ Case
Michigan Attorney General Dana Nessel filed charges last year against 16 alternate electors from the 2020 election.


Dairy Cows Must Be Tested for Bird Flu Before Moving Between States: USDA
New order announced Wednesday.


Chinese National Sentenced to 9 Months in Prison for Threatening Pro-Democracy Activist
‘The defendant’s crimes are serious. He weaponized the authoritarian nature of the PRC government in order to harass and threaten Ms. Zoey,’ said prosecutors.


Trump Media Asks Congress to Investigate Market Manipulation Claims
The CEO of the company, which owns Truth Social, raised the ’troubling' concerns.


Supreme Court Seems Sympathetic to Idaho’s Claim Its Strict Abortion Ban Is Valid
The issue is whether a federal law against ‘patient dumping’ creates a backdoor exception to the state’s strict abortion law.


Democrat House Representative Donald Payne Dies at 65
New Jersey governor Phil Murphy confirmed the development on Wednesday.




Biden Administration Sued Over ‘Blatant Power Grab’ by Banning Worker Noncompete Agreements
The plaintiff called the ban ‘not only unlawful but also a blatant power grab that will undermine American businesses’ ability to remain competitive.’


Inside the Pro-Palestinian Occupation of Columbia University
It was Day 6 of the ‘Gaza Solidarity Encampment,’ with hundreds of people, including students, sleeping in tents on the lawn of Columbia University’s campus.


Supreme Court May Rule for Starbucks in Labor Organizing Dispute
The coffeehouse chain is fighting a lower court order forcing it to rehire labor organizers whom Starbucks claims violated company policy.



Editors' Picks




Judge Unseals Documents Showing FBI Discussed ‘Loose Surveillance’ of Trump’s Plane
A large tranche of documents were unsealed by Judge Aileen Cannon on Monday, revealing the FBI’s code name for the probe.



The Return of Carob—This Time as a Functional Food
High in fiber and minerals and low in fat, carob—hailed as a “health food” in the 1970s—is making a comeback.


Business Advocates Argue California’s ‘Right to Disconnect’ Bill Is Overbroad
The groups said the bill ignores existing labor laws and would impact the ability of some offices to operate efficiently.


Police Officer Disciplined Over Undercover Conduct on Jan. 6: Agency
One document says the officer ‘participated’ in the riot at the U.S. Capitol.



Mahler’s ‘Resurrection’ Symphony: Answering Nihilism
Mahler’s “Resurrection” Symphony is the musical equivalent of “Hamlet.” What led to its creation?
Mahler’s ‘Resurrection’ Symphony: Answering Nihilism
Mahler’s “Resurrection” Symphony is the musical equivalent of “Hamlet.” What led to its creation?

Epoch Readers’ Stories
A Nation Divided
Poem by an American Patriot
What Is Going on Here?
There are two major things plants need to survive and continue generating our life saving oxygen. The first is CO2, and the second is sunshine.
Wisdom From a Retired Cowboy Artist
I have lived the life of the sculptures I have made.
Inspired Stories
Empower the World with Your Story: Share Love, Inspiration, and Hope with Millions


Special Coverage
Special Coverage































































Epoch Health

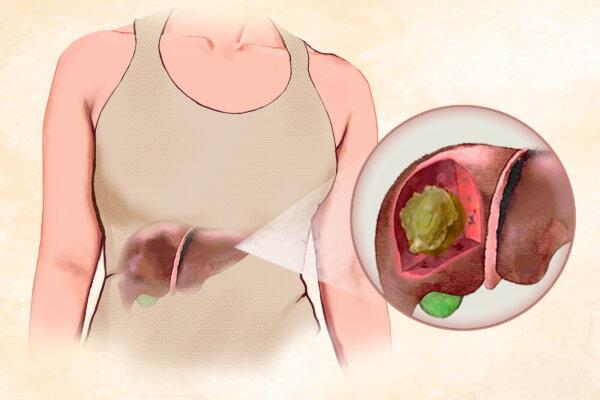
Atrial Fibrillation More Common and Risky in People Under 65 Than Previously Thought: Study
More than 25 percent of patients studied were under 65. Previously, it was thought that roughly 2 percent of people in this age group had Afib.

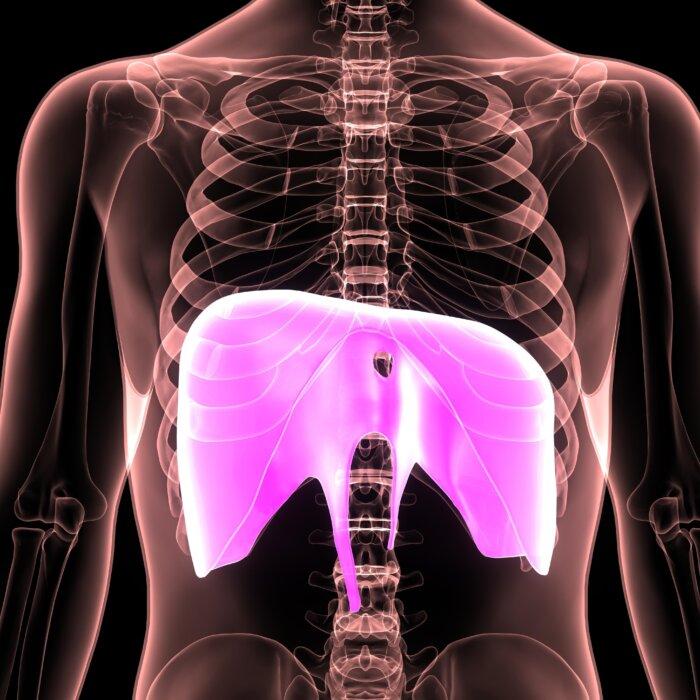
Atrial Fibrillation More Common and Risky in People Under 65 Than Previously Thought: Study
More than 25 percent of patients studied were under 65. Previously, it was thought that roughly 2 percent of people in this age group had Afib.
Bright

Palazzo del Te: A Palace Near Mantua, Italy
In this installment of ‘Larger Than Life: Architecture Through the Ages,’ we visit a duke’s ‘pleasure palace.’

Robert Smalls: Navy Captain and Reconstruction-Era Politician
This former slave would not let anything stop him on the road to freedom.

How Art Is Helping Veterans With PTSD
Artist Tim Gagnon gives veterans a way to move forward in civilian life.

Original ‘Naked Gun’ Director David Zucker: ‘To make fun of the left, you really can’t do that in Hollywood’
A master of the spoof genre, Mr. Zucker says studio executives are ‘overly sensitive.’

Jedidiah Morse: Father of American Geography
In this installment of ‘Profiles in History,’ we meet a minister who possessed a keen interest in geography and a concern about Christian liberalism.

How the US–Iraq Relationship Devolved Into War
Decades of animosity swirl in Steve Coll’s ‘The Achilles Trap: Saddam Hussein, the C.I.A., and the Origins of America’s Invasion of Iraq.'
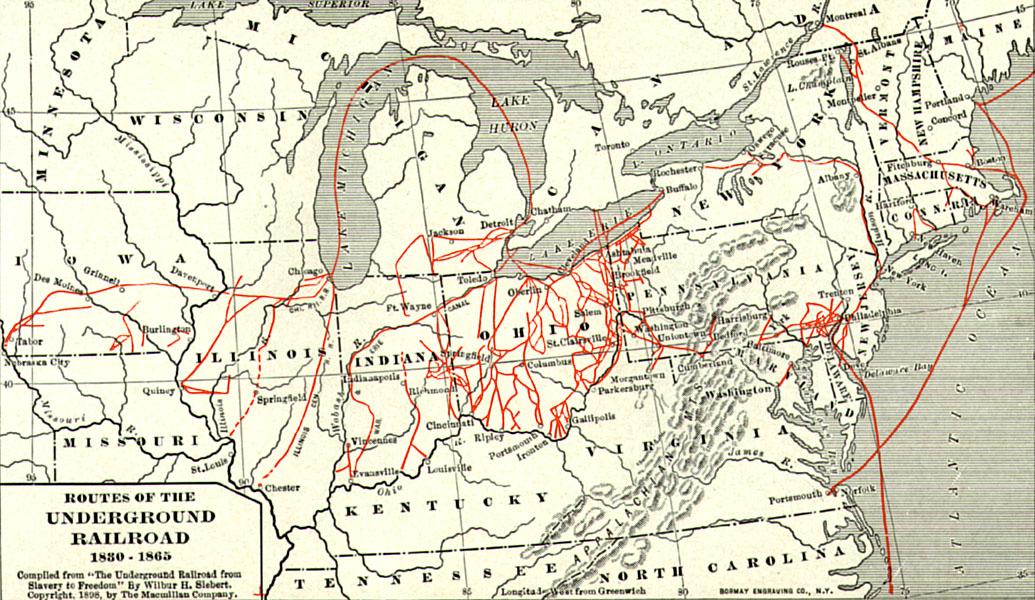
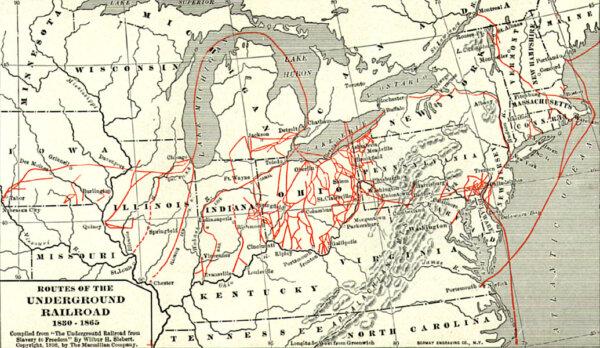
Important Players in a Divided Nation
Sons of the North, including young Spencer Kellogg, gave their all for the fight against slavery during the Civil War.








Palazzo del Te: A Palace Near Mantua, Italy
In this installment of ‘Larger Than Life: Architecture Through the Ages,’ we visit a duke’s ‘pleasure palace.’









You Need a Kitchen Assistant
Your slow cooker can help you prepare a meal for less that two cents an hour.












Rocky Mountaineer Train Launches Summer Season Promising Spectacular Views
Rocky Mountaineer has four routes that will take passengers through Western Canada and the American Southwest.

After 5 Years of Closure, ‘Glamping’ Back Again in Yosemite National Park
Camping hopefuls can now enter a lottery to experience three of the five available campsites.

Ed Perkins on Travel: Solo Travel—Difficult but Improving
The travel industry is slowly making prices fair for true solo travelers.

Rocky Mountaineer Train Launches Summer Season Promising Spectacular Views
Rocky Mountaineer has four routes that will take passengers through Western Canada and the American Southwest.









